

Novel Stem Cell-Derived Model Created of Inflammatory Neurological Disorder
Brain-in-a-dish approach also showed two existing HIV drugs may be effective in rescuing mutated cells
Published Date
By:
- Scott LaFee
Share This:
Article Content
An international team of scientists, led by University of California San Diego School of Medicine researchers, has created a human stem cell-based model of a rare, but devastating, inherited neurological autoimmune condition called Aicardi-Goutieres Syndrome (AGS). In doing so, the team was able to identify unusual and surprising underlying genetic mechanisms that drive AGS and test strategies to inhibit the condition using existing drugs.
Two repurposed FDA-approved drugs showed measurable effect, rescuing cells from the effects of AGS. The findings point to the promise of future clinical trials and to the utility of creating novel stem cell-based models of human diseases when no other models are available.
The findings are published in the August 10 online issue of Cell Stem Cell.
“Our approach can now be used to investigate other neurological conditions, like autism and schizophrenia and overlapping autoimmune disorders that dysfunction in similar ways,” said Alysson Muotri, PhD, professor in the UC San Diego School of Medicine departments of Pediatrics and Cellular and Molecular Medicine, director of the UC San Diego Stem Cell Program and a member of the Sanford Consortium for Regenerative Medicine.
First described in 1984, AGS typically involves early-onset inflammation affecting the brain, immune system and skin. Its severity depends upon which genes are involved — there are six types — but usually results in pronounced physiological and psychological consequences, from microcephaly (an abnormally small head) and spasticity to skin and vision problems and joint stiffness, all appearing in the first year of life. The syndrome is progressive, resulting in death or a persistent vegetative state in early childhood. Currently, there is no cure; the only treatments are symptomatic or palliative.
The clinical features of AGS mimic those of viral infections acquired in utero, before birth, with increased levels of inflammatory markers and other signatures of inflammatory response. However, Muotri said there is no link between AGS and exogenous pathogens. Previous research has shown that AGS patients have mutations in genes critical to nucleic acid metabolism in the regulation of cellular immune response, among them a deficiency in an enzyme called TREX1, which helps prevent abnormal DNA from accumulating in cells.
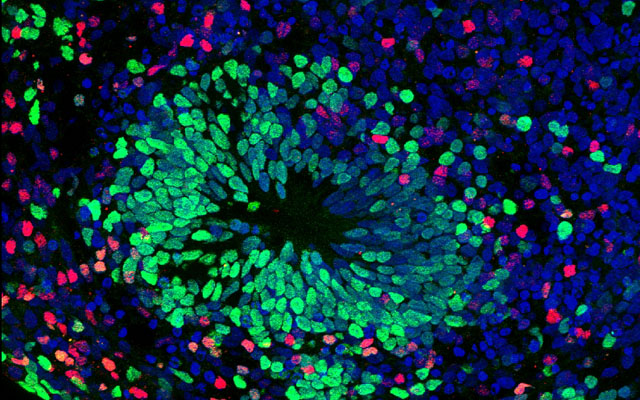
Deeper probing into the pathogenesis of AGS has been difficult because animal models do not accurately mimic the human version of the disease. So Muotri, with colleagues, used embryonic stem cells and induced pluripotent stem cells (iPSCs) derived from AGS patients to create six cellular models of the condition. In the past, Muotri’s lab has developed similar “disease-in-a-dish” neuronal models of autism, anorexia nervosa and Williams Syndrome, among other rare genetic neurological conditions.
From the iPSCs, they also created cerebral organoids or “mini-brains” — larger clusters of neurons that organize themselves into a cortical structure, similar to a developing human cerebral cortex.
The researchers found that with TREX1 not functioning normally, all of the cell models displayed excess extra-chromosomal DNA and that a major source of the excess DNA came from LINE1 (L1) retroelements. L1s are repetitive sequences of DNA with the ability to autonomously copy-and-paste themselves within the human genome. In the past, they have been called “jumping genes” and, because their function within cells is largely unknown, “junk DNA.”
However the term “junk DNA” is increasingly becoming a misnomer. In work published in 2005, for example, Muotri and colleagues reported that L1s have a high impact on brain cells compared to other tissues, suggesting an important, if so far mysterious, role in brain development.
Since then, he said, researchers around the world have investigated the role of L1s in creating a genetic mosaicism in the brain. “These are ancient, genomic parasites that replicate inside our cells. The majority of the current work is focusing on the impact of this genome mosaicism, but we decided to also look outside of the nucleus. And what we found was a big surprise.”
In some of the AGS cell models created by the researchers, toxins from excess DNA built up. Others showed an abnormal immune response, secreting toxins that induced cell death in other cells. The combined effect in organoids was a massive reduction in neuron growth when the opposite should occur. “These models seemed to mirror the development and progression of AGS in a developing fetus,” said Muotri. “It was cell death and reduction when neural development should be rising.”
The cell death was trigged by the anti-viral response from the L1 molecules outside the nucleus. “We uncovered a novel and fundamental mechanism, where chronic response to L1 elements can negatively impact human neurodevelopment,” said Charles Thomas, a former graduate student in the Muotri lab and first author of the study. “This mechanism seems human-specific. We don’t see this in the mouse.”
The researchers observed that AGS pathogenesis was similar to a retroviral infection and wondered whether existing HIV antiretroviral drugs might be effective in interfering in L1 replication. Two drugs were tested in the cell models: Stavudine and Lamivudine. Both drugs resulted in reduced L1 and cell toxicity. Cell model growth returned in all cell types and in the complex, differentiated colonies of nerve cells that comprise organoids.
The data supported the idea that HIV drugs could benefit AGS patients, Muotri said. A clinical trial led by study co-author Yanick Crow, MRCP, PhD, at Sorbonne Paris Cite University and the University of Manchester, has already started in Europe.
Muotri said the findings were illuminating and encouraging, providing a platform and impetus for further study of the pathology of neuroinflammation and drug discovery. “It’s important to note that while this work focused on AGS, nerve cells in schizophrenia show an overabundance of L1 elements — and there is an overlap with other autoimmune disorders.
“This is a great example of how a fundamental basic research could be rapidly translated into clinics. Are there analogous mechanisms at work in these different diseases? Is this modeling strategy relevant for better understanding and treating them? These are questions we will now pursue.”
Co-authors of this study include: Leon Tejwani, Cleber A. Trujillo, Priscilla D. Negraes, Roberto H. Herai, Pinar Mesci, and Angela Macia, UC San Diego; Yanick J. Crow, Sorbonne Paris Cite University, Institut Imagine, Hopital Necker, Paris and University of Manchester, UK.
Funding for this research came, in part, from the National Institutes of Health (R01MH108528, R01MH094753, R01MH109885, R01MH100175, R21MH107771, R56MH109587, U19MH107367), the Ruth L. Kirschstein NRSA Fellowship (1F31NS076198), the California Institute for Regenerative Medicine (DISC1-08825), UC San Diego Altman Clinical and Translational Research Institute, NARSAD, the European Research Council (GA 309449), ERA-NET Neuron (MR/M501803/1) and the National Research Agency of France (ANR-10-IAHU-01).
Share This:
You May Also Like


Stay in the Know
Keep up with all the latest from UC San Diego. Subscribe to the newsletter today.