
Scientists Discover a New Signaling Pathway and Design a Novel Drug for Liver Fibrosis
Health & Behavior
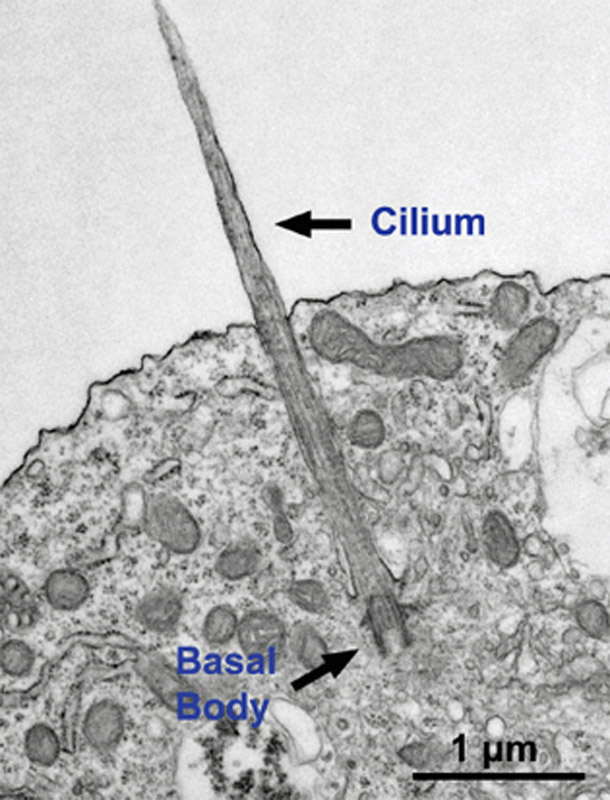
Image using an electron microscope shows a cilium growing from a neuron. -Gleeson lab, UC San Diego
A team led by researchers at the University of California, San Diego School of Medicine reports that newly discovered mutations in an evolved assembly of genes cause Joubert syndrome, a form of syndromic autism.
The findings are published in the January 26 online issue of Science Express.
Joubert syndrome is a rare, recessive brain condition characterized by malformation or underdevelopment of the cerebellum and brainstem. The disease is due specifically to alterations in cellular primary cilia – antenna-like structures found on most cells. The consequence is a range of distinct physical and cognitive disabilities, including poor muscle control, and mental retardation. Up to 40 percent of Joubert syndrome patients meet clinical criteria for autism, as well as other neurocognitive disorders, so it is considered a syndromic form of autism.
The cause or causes of Joubert syndrome are not well-understood. Researchers looked at mutations in the TMEM216 gene, which had previously been linked to the syndrome. However, only half of the expected Joubert syndrome patients exhibit TMEM216 gene mutations; the other half did not. Using genomic sequencing, the research team, led by Joseph G. Gleeson, MD, professor of neurosciences and pediatrics at UC San Diego, broadened their inquiry and discovered a second culprit: mutations in a neighboring gene called TMEM138.
“It is extraordinarily rare for two adjacent genes to cause the same human disease,” said Gleeson. “The mystery that emerged from this was whether these two adjacent, non-duplicated genes causing indistinguishable disease have functional connections at the gene or protein level.”
Through evolutionary analysis, the scientists concluded that the two TMEM genes became joined end-to-end approximately 260 million years ago, about the time some amphibians began transitioning into land-based reptiles. The connected genes evolved in tandem, becoming regulated by the same transcription factors.
“Prior to this transition, the two genes had wildly different expression levels,” said Jeong Ho Lee, MD, PhD, and first author of the study. “Following this transition, they became tightly co-regulated. Moreover, we found that the two encoded proteins coordinate delivery of factors key for cilia assembly.”
Gleeson said the findings suggest the human genome has evolved to take advantage of fortuitous ancestral events like gene translocations to better coordinate gene expression by assembling into specific modules. When these modules are disrupted, however, neurodevelopmental diseases may result.
This research was funded, in part, by the Simons Foundation, the American Philosophical Society, the National Institutes of Health and the Howard Hughes Medical Institute.
Co-authors are Jennifer L. Silhavy, Ji Eun Lee, Lihadh Al-Gazali, Sophie Thomas, Erica E Davis, Stephanie L. Bielas, Kiley J. Hill, Miriam Iannicelli, Francesco Brancati, Stacey B. Gabriel, Carsten Russ, Clare V. Logan, Saghira Malik Sharif, Christopher P. Bennett, Masumi Abe, Friedhelm Hildebrandt, Bill H. Diplas, Tania Attié-Bitach, Nicholas Katsanis, Anna Rajab, Roshan Koul, Laszlo Sztriha, Elizabeth R. Waters, Susan Ferro- Novick, Geoffrey C. Woods, Colin A. Johnson, Enza Maria Valente, Maha S. Zaki.
Keep up with all the latest from UC San Diego. Subscribe to the newsletter today.

