

Severe Newborn Jaundice Could be Preventable, Mouse Study Shows
Enzyme rapidly breaks down bilirubin when researchers inhibit intestinal protein NCoR1
Published Date
By:
- Heather Buschman, PhD
Share This:
Article Content
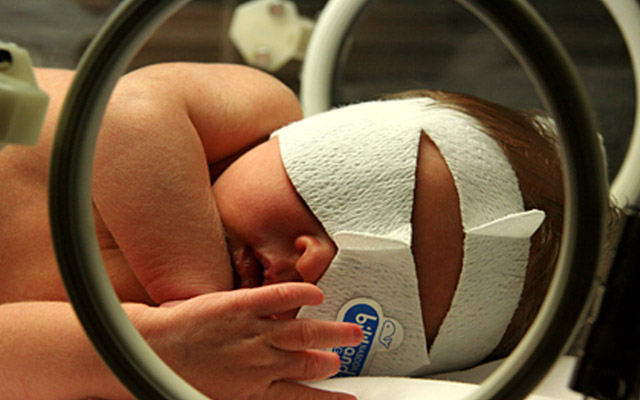
For many newborn babies, an enzyme that breaks down the molecule bilirubin doesn’t activate right away. The resulting bilirubin buildup can lead to jaundice, a typically harmless condition that causes a baby’s skin to temporarily appear yellow. In some cases, however, bilirubin can accumulate to toxic levels in the brain. Researchers at University of California San Diego School of Medicine have identified a protein that inhibits the bilirubin-breakdown enzyme. Methods that block this inhibitor, and thus restore the enzyme’s activity, could provide a new therapeutic approach for preventing or treating severe jaundice.
The study is published February 6 by the Proceedings of the National Academy of Sciences.
“This is the first report that describes the molecular processes that dictate the onset and control of the most medically worrisome form of jaundice in newborns, a condition known as severe neonatal hyperbilirubinemia,” said study co-author Robert Tukey, PhD, professor of pharmacology at UC San Diego School of Medicine. “This new information will help us look for drugs or dietary therapeutics that alleviate the early onset of bilirubin toxicity.”
At birth, newborns are suddenly exposed to unprecedented levels of oxygen, resulting in the rapid but temporary destruction of red blood cells and spillage of excess bilirubin in the bloodstream. If not properly broken down by an enzyme called UDP-glucuronosyltransferase 1A1 (UGT1A1), bilirubin continues to accumulate. High bilirubin levels in the brain can lead to encephalopathy, seizures, life-long brain damage and even death.
To better understand UGT1A1’s role in human newborns, Tukey’s collaborator and senior author Shujuan Chen, PhD, assistant professor of pharmacology at UC San Diego School of Medicine, replaced the native UGT1A1 gene in mice with the human version of the gene. While normal mice don’t develop jaundice at birth, the researchers found that “humanized” mice developed severe neonatal hyperbilirubinemia and some of the resulting health consequences.
Tukey, Chen and team also discovered that the UGT1A1 gene is turned off in liver tissue in newborn humanized mice, as in humans, but also repressed in the gastrointestinal tract. They eventually identified the cause of UGT1A1’s inhibition in humanized newborn mice — a repressor protein called nuclear corepressor protein 1 (NCoR1).
When the researchers deleted the NCoR1 gene from the mice’s intestinal tissue, the UGT1A1 gene was activated. Newly restored UGT1A1 broke down the excess bilirubin, eliminating signs of severe neonatal hyperbilirubinemia in the humanized mice.
“Since we now know that intestinal tissue is at least partly responsible for regulating bilirubin toxicity, we’re hopeful that oral therapeutics could be developed to block the onset of severe neonatal hyperbilirubinemia,” said Chen.
In countries with adequate health care systems, severe neonatal hyperbilirubinemia can be managed with phototherapy and blood transfusions. However, in many parts of the world, such as sub-Saharan Africa, South Asia and other places where preterm births are on the rise, rapid bilirubin rise often goes untreated. Each year, more than 1 million newborns worldwide experience severe neonatal hyperbilirubinemia.
Study co-authors include: Wenqi Lu, Mei-Fei Yueh, Eva Rettenmeier, Miao Liu, Kepeng Wang, Michael Karin, UC San Diego; Johan Auwerx, Ecole Polytechnique Fédérale de Lausanne; Ruth T. Yu, and Ronald M. Evans, Howard Hughes Medical Institute and Salk Institute for Biological Studies.
This research was funded, in part, by the National Institutes of Health (ES010337, GM086713, GM100481, R21ES024818, AI043477) and Swiss National Science Foundation (310030B-160318).
Share This:


Stay in the Know
Keep up with all the latest from UC San Diego. Subscribe to the newsletter today.