

Why Don’t We All Get Alzheimer’s Disease?
By:
- Scott LaFee
Published Date
By:
- Scott LaFee
Share This:
Article Content
Though one might think the brains of people who develop Alzheimer’s disease (AD) possess building blocks of the disease absent in healthy brains, for most sufferers, this is not true. Every human brain contains the ingredients necessary to spark AD, but while an estimated 5 million Americans have AD – a number projected to triple by 2050 – the vast majority of people do not and will not develop the devastating neurological condition.
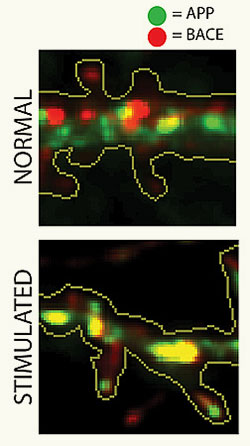
Top: Vesicles containing APP (green) and BACE (red) are normally segregated in neurons. Bottom: After neuronal stimulation, known to produce more beta-amyloid, APP and BACE converge in common vesicles, depicted in yellow.
For researchers like Subhojit Roy, MD, PhD, associate professor in the Departments of Pathology and Neurosciences at the University of California, San Diego School of Medicine, these facts produce a singular question: Why don’t we all get Alzheimer’s disease?
In a paper published in the August 7 issue of the journal Neuron, Roy and colleagues offer an explanation – a trick of nature that, in most people, maintains critical separation between a protein and an enzyme that, when combined, trigger the progressive cell degeneration and death characteristic of AD.
“It’s like physically separating gunpowder and match so that the inevitable explosion is avoided,” said principal investigator Roy, a cell biologist and neuropathologist in the Shiley-Marcos Alzheimer’s Disease Research Center at UC San Diego. “Knowing how the gunpowder and match are separated may give us new insights into possibly stopping the disease.”
The severity of AD is measured in the loss of functioning neurons. In pathological terms, there are two tell-tale signs of AD: clumps of a protein called beta-amyloid “plaques” that accumulate outside neurons and threads or “tangles” of another protein, called tau, found inside neurons. Most neuroscientists believe AD is caused by the accumulating assemblies of beta-amyloid protein triggering a sequence of events that leads to impaired cell function and death. This so-called “amyloid cascade hypothesis” puts beta-amyloid protein at the center of AD pathology.
Creating beta-amyloid requires the convergence of a protein called amyloid precursor protein (APP) and an enzyme that cleaves APP into smaller toxic fragments called beta-secretase or BACE.
“Both of these proteins are highly expressed in the brain,” said Roy, “and if they were allowed to combine continuously, we would all have AD.”
But that doesn’t happen. Using cultured hippocampal neurons and tissue from human and mouse brains, Roy – along with first author Utpal Das, a postdoctoral fellow in Roy’s lab, and colleagues – discovered that healthy brain cells largely segregate APP and BACE-1 into distinct compartments as soon as they are manufactured, ensuring the two proteins do not have much contact with each other.
“Nature seems to have come up with an interesting trick to separate co-conspirators,” said Roy.
The scientists also found that the conditions promoting greater production of beta-amyloid protein boost the convergence of APP and BACE. Specifically, an increase in neuronal electrical activity – known to increase the production of beta-amyloid – also led to an increase in APP-BACE convergence. Post-mortem examinations of AD patients revealed increased physical proximity of the proteins as well, adding support to the pathophysiological significance of this phenomenon in human disease.
Das said the findings are fundamentally important because they elucidate some of the earliest molecular events triggering AD and show how a healthy brain naturally avoids them. In clinical terms, they point to a possible new avenue for ultimately treating or even preventing the disease.
“An exciting aspect is that we can perhaps screen for molecules that can physically keep APP and BACE-1 apart,” said Das. “It’s a somewhat unconventional approach.”
Co-authors are David Scott, Archan Ganguly and Yong Tang, UCSD Departments of Pathology and Neurosciences; and Edward H. Koo, UCSD Department of Neurosciences. Roy and Koo are also members of the Shiley-Marcos Alzheimer’s Disease Research Center (ADRC) at UC San Diego.
Funding for this research came from the American Federation for Aging Research, National Institutes of Health grant P50AG005131 and a gift from Darlene Shiley to the ADRC.
Share This:
You May Also Like


Stay in the Know
Keep up with all the latest from UC San Diego. Subscribe to the newsletter today.