

A Quantum Leap for Molecular Simulations on GPUs
Researchers develop open-source software to ease computational science on modern computers
Published Date
By:
- Cynthia Dillon
Share This:
Article Content
Developing improved materials for things such as energy storage and drug discovery is of interest to researchers and society alike. Quantum mechanics (QM), a theory that describes the physical properties of nature on the atomic and subatomic scale, is the basis for molecular and materials scientists who develop these useful, futuristic products.
The challenge is that the QM calculations to describe the many properties of molecules and the materials they make up require a lot of computer power.
This is where a small team of postdoctoral scholars led by Andreas Goetz of the San Diego Supercomputer Center (SDSC) at UC San Diego and his colleague Kenneth Merz at Michigan State University (MSU) has developed software that takes advantage of powerful graphics processing units (GPUs) for these complex QM calculations of molecules.
Their research was recently featured in two peer-reviewed publications: the Journal of Chemical Theory and Computation (JCTC) and the Journal of Chemical Information and Modeling (JCIM).
“The need of freely available open-source software to enable computational molecular sciences on modern computers—from desktops to supercomputers with thousands of GPUs—inspired our efforts,” said Goetz, director of the Computational Chemistry Laboratory at SDSC. “This research provides open-source software for efficient QM calculations and for mixed quantum/classical (QM/MM) simulations of molecular systems on graphics processing units.”
The software enables researchers to address problems that would otherwise be computationally intractable in a range of research areas such as computational materials design, catalyst design and drug design.
Moreover, Merz notes, “Our code can also be used in a high-throughput fashion to generate data for Artificial Intelligence-based molecular design approaches, such as developing improved materials and drugs.”
Merz’ group at MSU and Goetz’s SDSC group jointly designed the research, developed algorithms and engineered the software. The scientists used data collected on SDSC’s Expanse supercomputer.
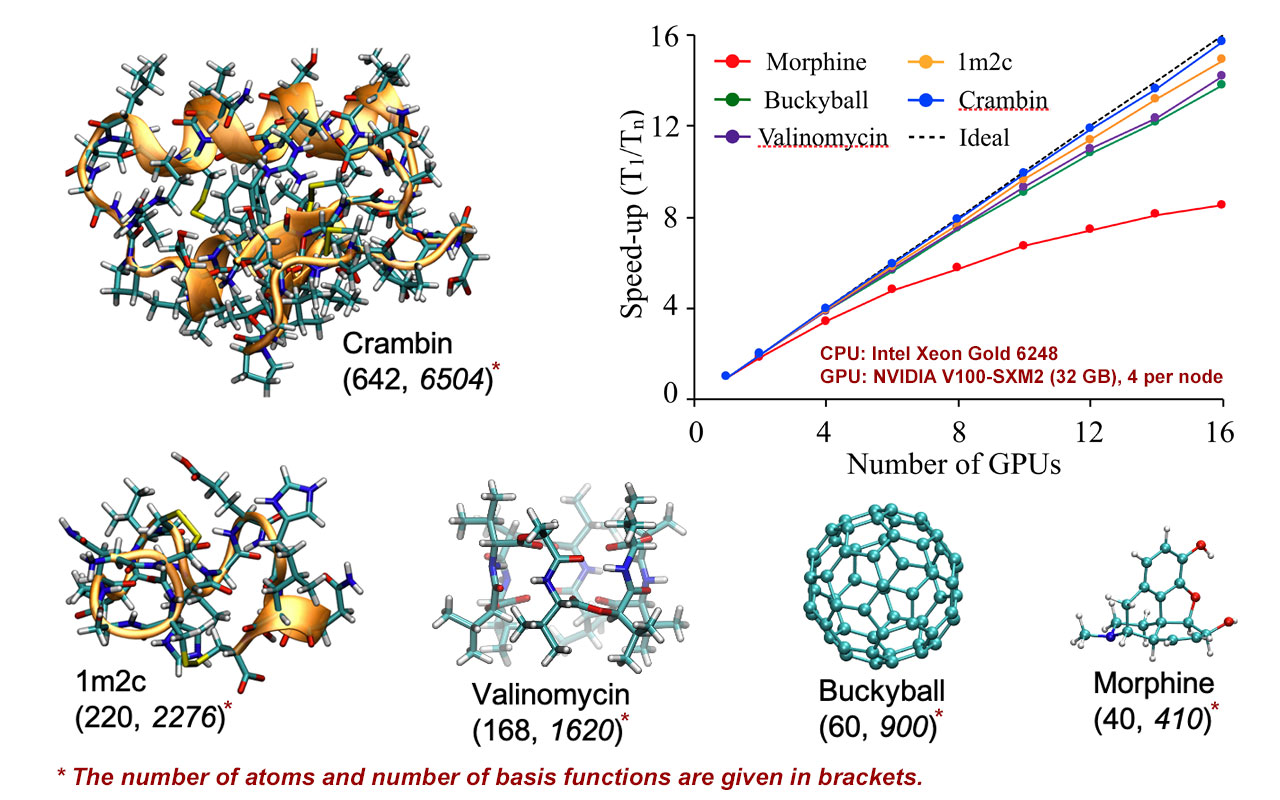
In their JCTC paper, “Harnessing the Power of Multi-GPU Acceleration into the Quantum Interaction Computational Kernel Program,” the researchers explain supercomputers in a simplified way as made up of many tightly integrated computers that work in parallel together to solve complex computational problems. So, their software, called QUICK, coordinates computations across multiple GPUs to get solutions more quickly into the hands of researchers.

Artistic representation of the novel integration of the QUantum Interaction Computational Kernel (QUICK) program for multi-GPU enabled electronic structure calculations in QM/MM simulations with the AMBER molecular dynamics software package. A relevant portion of a protein is computed with QM while the surrounding region is represented via a classical MM model. (Image credit: V. W. D. Cruzeiro, A. W. Goetz, B. Tolo)
“This is like coordinating a fleet of trucks to deliver time-sensitive goods quickly from one place to another,” explained Madushanka Manathunga, postdoctoral scholar at MSU. “We had to dissect the mathematical equations underlying the QM models to understand how we can efficiently parallelize the computations and effectively distribute the work onto multiple GPUs.”
In their JCIM paper, “Open-Source Multi-GPU-Accelerated QM/MM Simulations with AMBER and QUICK,” the researchers consider their QM/MM coupling, which uses their faster GPU code for the complex QM calculations of, in this case, an active site of an enzyme together with a simplified MM model for the remainder of the protein beyond the active site region.
The importance of such computational methods has been recognized with the award of the 2013 Nobel Prize in chemistry for the development of multi-scale models of complex chemical systems.
“It’s exciting to have this multi-GPU QM code QUICK available for QM/MM simulations within the AMBER suite of programs so the community can take advantage of our code base to add new features and to use it to solve a range of chemical and biological problems,” Merz said.
Vinícius Cruzeiro, postdoctoral scholar at SDSC/UC San Diego, who co-led the software engineering work and co-wrote the papers with Manathunga, describes their technical work.
“This is like using a ‘computational microscope’ to zoom in on very tiny objects, such as important parts of a spike protein on a virus, to further our understanding of what is going on in detail and provide crucial information that might not be accessed experimentally,” he said.
According to the researchers, their software enables so-called ab initio Hartree-Fock and density functional theory (DFT) QM calculations. Due to its favorable ratio of computational cost to accuracy, DFT is widely used in computational molecular sciences.
“The features implemented in QUICK are available in commercial programs, but these require costly license fees, Cruzeiro said. “Therefore, it is rewarding to work in a project that aims to bring these features free-of-charge to researchers all over the world, especially to those impacted by paywalls."
Implementing their software on GPUs written in a special programming language, the researchers used the message passing interface (MPI) to distribute work onto different GPUs both within and across compute nodes. Part of this work was initiated last year at the first digital GPU Hackathon co-hosted by SDSC and Nvidia.
“All of this work was done remotely due to the Covid-19 pandemic, but this is not a big problem for software-related work,” Goetz said.
Other contributors included Kristopher Keipert and Kamesh Arumugam from N, Chi Jin from MSU, Yipu Miao from Facebook and a former MSU graduate student, Metin Aktulga from MSU and Dawei Mu from NCSA, formerly at SDSC.
“I love working with smart people on challenging problems, and I love teaching computers how to solve our science problems,” Goetz said.
This work was supported by the National Science Foundation (NSF) (OAC-1835144); San Diego Supercomputer Center (computer time); the Department of Chemistry and Biochemistry and High-Performance Computer Center (iCER HPCC) at MSU; Nvidia (access to the Nvidia Cluster). This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF (ACI1053575, TG-CHE130010).
Share This:


Stay in the Know
Keep up with all the latest from UC San Diego. Subscribe to the newsletter today.