

Breaking Bad Mitochondria
Mechanism helps explain persistence of hepatitis C virus
Published Date
By:
- Scott LaFee
Share This:
Article Content
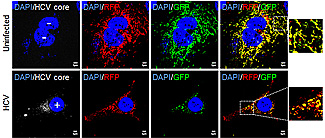
Mitochondria in hepatitis C-infected cells (bottom row) are self-destructing. The self-annihilation process explains the persistance and virulence of the virus in human liver cells.
Researchers at the University of California, San Diego School of Medicine have identified a mechanism that explains why people with the hepatitis C virus get liver disease and why the virus is able to persist in the body for so long.
The hard-to-kill pathogen, which infects an estimated 200 million people worldwide, attacks the liver cells’ energy centers – the mitochondria – dismantling the cell’s innate ability to fight infection. It does this by altering cells mitochondrial dynamics.
The study, published in today’s issue of the Proceedings of the National Academy of Sciences, suggests that mitochondrial operations could be a therapeutic target against hepatitis C, the leading cause of liver transplants and a major cause of liver cancer in the U.S.
“Our study tells us the story of how the hepatitis C virus causes liver disease,” said Aleem Siddiqui, PhD, professor of medicine and senior author. “The virus damages mitochondria in liver cells. Cells recognize the damage and respond to it by recruiting proteins that tell the mitochondria to eliminate the damaged area, but the repair process ends up helping the virus.”
Mitochondria are organelles in a cell that convert energy from food (glucose) into a form of energy that can be used by cells called adenosine triphosphate.
Specifically, the researchers discovered that the virus stimulates the production of a protein (Drp 1) that induces viral-damaged mitochondria to undergo asymmetric fragmentation. This fragmentation (fission) results in the formation of one healthy mitochondrion and one damaged or bad mitochondrion, the latter of which is quickly broken down (catabolized) and dissolved in the cell’s cytoplasm.
Although the fragmentation serves to excise the damaged area from the mitochondrion, the formation of a healthy mitochondrion also helps keep the virus-infected cell alive. Moreover, the virus is able to use the mitochondrial remains (all the amino acids and lipids from the catabolized mitochondrion) to help fuel its continued replication and virulence.
“It’s like the bad part of the house is demolished to the benefit of the virus,” Siddiqui said.
In their experiments, the researchers showed that hepatitis C-infected cells with higher Drp 1 protein levels also produced less interferon, the body’s natural immune booster. These cells were also less likely to undergo apoptosis, a process that would encourage damaged cells to essentially kill themselves.
The reverse was also observed: When the Drp 1 protein was “silenced,” interferon production and apoptotic activity increased.
“Mitochondrial processes are at the center of understanding the persistence of the virus and how it flies under the radar of the body’s natural immune response,” he said. “The trick is to find a way to deliver a drug that could target the Drp 1 protein specifically in hepatitis C-infected liver cells, maybe through nanotechnology.”
Co-authors include Seong-Jun Kim and Gulam H. Syed, Mohsin Khan and Wei-Wei Chiu Division of Infectious Diseases, UCSD; Muhammad A. Sohail, Division of Gastroenterology, UCSD; and Robert G. Gish, Hepatitis B Foundation.
This research was funded, in part, by National Institutes of Health grants AI085087, DK077704, DK08379 and T32 DK07202.
Share This:
You May Also Like


Stay in the Know
Keep up with all the latest from UC San Diego. Subscribe to the newsletter today.