

How SARS-CoV-2 Hijacks Human Cells to Evade Immune System
Published Date
By:
- Heather Buschman, PhD
Share This:
Article Content
Researchers at University of California San Diego School of Medicine have discovered one way in which SARS-CoV-2, the coronavirus that causes COVID-19, hijacks human cell machinery to blunt the immune response, allowing it to establish infection, replicate and cause disease.
In short, the virus’ genome gets tagged with a special marker by a human enzyme that tells the immune system to stand down, while at the same time ramping up production of the surface proteins that SARS-CoV-2 uses as a “doorknob” to enter cells.
The study, published April 22, 2021 in Cell Reports, helps lay the groundwork for new anti-viral immunotherapies — treatments that work by boosting a patient’s immune system, rather than directly killing the virus.
“It’s very smart of this virus to use host machinery to simultaneously go into stealth mode and get inside more cells,” said Tariq Rana, PhD, professor and chief of the Division of Genetics in the Department of Pediatrics at UC San Diego School of Medicine and Moores Cancer Center. “The more we know about how the virus establishes itself in the body, the better equipped we are to disrupt it.”
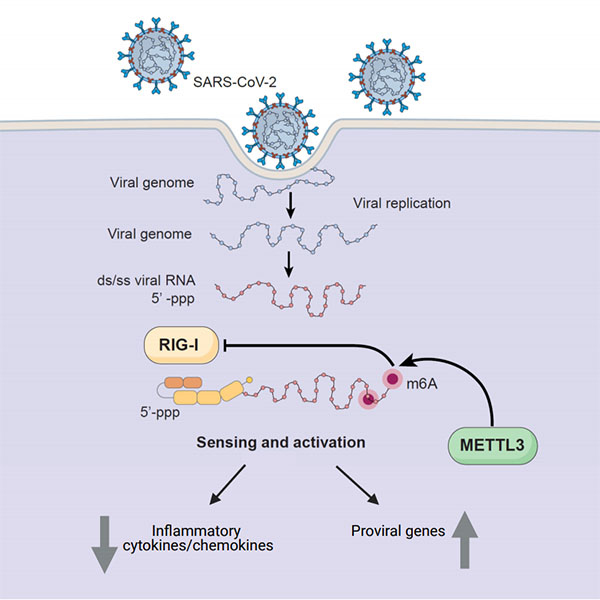
Human enzyme METTL3 adds methyl groups to introduce m6A in SARS-CoV-2’s RNA. That modification prevents the virus’ RNA from triggering inflammatory molecules known as cytokines. METTL3 also leads to increased expression of pro-viral genes — those that encode proteins needed for SARS-CoV-2 replication and survival.
In human cells, genes (DNA) are transcribed into RNA, which is then translated into proteins, the molecules that make up the majority of cells. But it’s not always so straightforward. Cells can chemically modify RNA to influence protein production. One of these modifications is the addition of methyl groups to adenosine, one of the building blocks that make up RNA. Known as N6-methyladenosine (m6A), this modification is common in humans and other organisms, including viruses.
In contrast to humans, the entire genomes of some viruses, including SARS-CoV-2, are made up of RNA instead of DNA. And rather than carry around the machinery to translate that into proteins, the coronavirus gets human cells to do the work.
Rana and his team previously discovered that m6A plays an important role in HIV and Zika virus infections. In their latest study, the researchers discovered that the human enzyme METTL3 adds methyl groups to introduce m6A in SARS-CoV-2’s RNA. That modification prevents the virus’ RNA from triggering inflammatory molecules known as cytokines. To the team’s surprise, METTL3’s activity also led to increased expression of pro-viral genes — those that encode proteins needed for SARS-CoV-2 replication and survival, such as ACE2, the cell surface receptor that the virus uses to enter human cells.
“It remains to be seen why our cells help the virus out like this,” Rana said.
When the team removed METTL3 from cells in the laboratory, using gene silencing or other methods, they saw the reverse — a pro-inflammatory molecule known as RIG1 binds the viral RNA, more inflammatory cytokines were produced, and pro-viral genes were inhibited. Ultimately, inhibiting METTL3 suppressed viral replication.
To see how this mechanism plays out in the real world, the team compared post-mortem lung samples from COVID-19 patients and healthy lung biopsies. In patients who had died from severe COVID-19, the team found, METTL3 expression was lower and inflammatory genes were elevated. That makes sense in later stages of COVID-19, Rana said, because cytokine storm — the excessive activation of the patient’s own immune system — is known to worsen the disease.
“It’s like there are two phases of the infection — in the first, the virus needs METTL3 to help it evade the immune response,” he said, “but in the second phase, once the virus is replicating like crazy, it’s better to downregulate METTL3.”
Rana’s team is now validating their findings in animal models, and developing METTL3 inhibitors to test as potential experimental therapies for COVID-19.
“We hope that by manipulating m6A levels in the virus, we might be able to time the innate immune response in a way that benefits patients with COVID19, especially for the mild or moderate patients who haven’t developed cytokine storm,” Rana said. “The challenge is that cells have many other enzymes like METTL3, known as methyltransferases, so inhibiting it would need to be done very specifically, at a specific time.”
Co-authors of the study include: Na Li, Hui Hui, Bill Bray, Rob Knight, Davey Smith, Aaron F. Carlin, UC San Diego; Gwendolyn Michelle Gonzalez, Yinsheng Wang, UC Riverside; Mark Zeller, Kristian G. Anderson, Scripps Research.
Funding for this research came, in part, from the National Institutes of Health (grants K08 AI130381, CA177322, DA039562, DA046171 and AI125103), Burroughs Wellcome Fund USA and John and Mary Tu Foundation.
Disclosure: Tariq Rana is a founder of ViRx Pharmaceuticals and has an equity interest in the company. The terms of this arrangement have been reviewed and approved by the University of California San Diego in accordance with its conflict of interest policies.
Share This:
You May Also Like


Stay in the Know
Keep up with all the latest from UC San Diego. Subscribe to the newsletter today.